|
Research Ideas and Outcomes :
Research Article
|
|
Corresponding author: Victor Padilla-Sanchez (70padillasan@cua.edu)
Received: 08 Jan 2021 | Published: 15 Jan 2021
© 2021 Victor Padilla-Sanchez
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Padilla-Sanchez V (2021) SARS-CoV-2 Structural Analysis of Receptor Binding Domain New Variants from United Kingdom and South Africa. Research Ideas and Outcomes 7: e62936. https://doi.org/10.3897/rio.7.e62936
|
|
Abstract
SARS-CoV-2 has caused more than 80 million infections and close to 2 million deaths worldwide as of January 2021. This pandemic has caused an incredible damage to humanity being it medically and/or financially halting life as we know it. If it were not enough, the current virus is changing to a more deadly form because of the mutations that are arising on its genome. Importantly, two variants have emerged in recent months, one in United Kingdom and the other in South Africa that are more infectious and escape antibody binding. These two variants have mutations in the receptor binding domain of the spike glycoprotein namely N501Y (UK, SA), K417N (SA) and E484K (SA). Here, I present a structural analysis of spike glycoprotein bound to ACE2 (angiotensin converting enzyme 2) where the mutations have been introduced in silico showing the reason why these variants bind better to ACE2 receptors.
Keywords
SARS-CoV-2, United Kingdom, South Africa, coronavirus, variants, in silico, chimera, molecular dynamics, RBD, spike, mutations, computational biology, immunology
Introduction
New variants of SARS-CoV-2 present a particular risk of infecting humans which we are starting to see these days (Fig.

SARS-CoV-2 viral infection at atomic resolution. Counting eight viruses, each of which has spikes (big protrusions) and E membrane proteins (small protrusions) rainbow colored and a core in sienna color, this picture shows how the viruses approach the cell membrane (green). The ACE2 receptors are colored magenta. The field of view is 1 micrometer.
The goal of this structural analysis is to give an explanation why these mutations in RBD increase transmissibility and therefore device strategies for passive immunity and vaccination that previous approaches have not accounted for.
Material and methods
Structural analysis has been performed with UCSF Chimera software, Visual Molecular Dynamics (VMD) and PyMol. Molecular Dynamics has been performed with Nanoscale Molecular Dynamics (NAMD) with default parameters and implicit solvent. Mutations have been introduced in silico in Chimera and analyzed (
Results
In the United Kingdom variant (Fig.
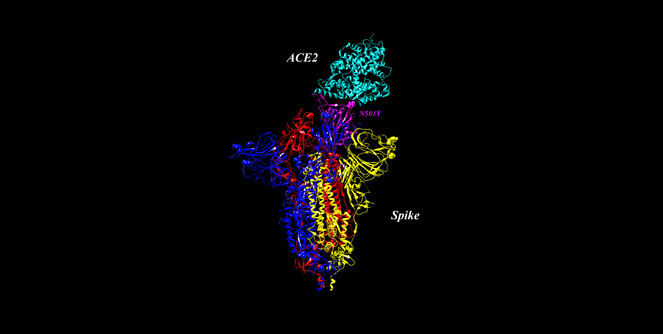
Spike glycoprotein bound to ACE2 receptor. PDB 7DF4 (
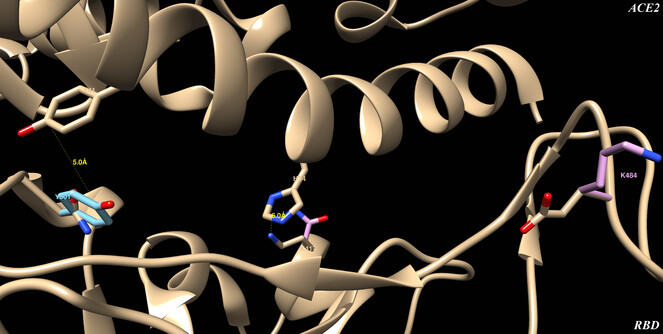
In the South African variant, besides N501Y we have K417N which provides less repulsive forces between ACE2-RBD since the wild type lysine is positive which is close to histidine (positive too) in ACE2 (H34). Changing to asparagine deletes this interaction therefore ACE2 can bind closer to the spike of the virus. Finally, E484K provides a lysine that serves as escape mutant against the immune system since interferes with the ability of antibodies to bind to this RBD region (Fig.
Discussion
The mutations in the spike glycoprotein that affect ACE2 receptor binding are N501Y for United Kingdom variant and two more, K417N and E484K for South Africa variant. The UK mutation interacts closely with Y41 in the receptor therefore producing aromatic-aromatic interactions that provide for stronger binding between receptor and spike.
On the other hand, the South Africa variant has two more mutations. K417N disrupts the repulsive forces between K417 and H34 (positive-positive) providing less repulsion between the binding partners which adds to the effect of Y501 aromatic attraction.
Finally, K484 has been recently demonstrated to be an escape mutant against the immune system since prevents binding from polyclonal human serum antibodies (
These mutations are spreading rapidly and more efficiently than the previous version of the virus therefore the public health concern is very critical. Further studies are needed to quantify the binding strength of these variants compared to wild type in wet lab experiments but the distance between Y501 and Y41 is very close, even when they move (molecular dynamics). Likewise, N417 does not interact anymore with H34 since this amino acid has a shorter side chain and not positive therefore the repulsive forces are disrupted between these two amino acids (K417-H34).
Due to the public health emergency that these variants are originating I found it URGENT to publish these results as soon as possible without further testing which the future will provide.
Acknowledgements
The author acknowledges the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing HPC and visualization resources that have contributed to the research results reported within this paper. URL: http://www.tacc.utexas.edu
References
-
Investigation of novel SARS-COV-2 variant: Variant of Concern 202012/01 (PDF) (Report). Public Health England.PHE.
-
Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies.bioRxivhttps://doi.org/10.1101/2020.12.31.425021
-
VMD: Visual molecular dynamics.Journal of Molecular Graphics14(1):33‑38. https://doi.org/10.1016/0263-7855(96)00018-5
-
UCSF Chimera--A visualization system for exploratory research and analysis.Journal of Computational Chemistry25(13):1605‑1612. https://doi.org/10.1002/jcc.20084
-
Scalable molecular dynamics on CPU and GPU architectures with NAMD.The Journal of Chemical Physics153(4). https://doi.org/10.1063/5.0014475
-
The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
-
Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM.Science Advances7(1). https://doi.org/10.1126/sciadv.abe5575
Supplementary materials
RBD-ACE2 complexes. In that order from left to right: wild type, United Kingdom mutant and South Africa mutant. Shown in sticks are the residues involved in interactions namely 501, 417, 484 in RBD (bottom) and Y41 and H34 in ACE2 (top).
RBD-ACE2 interface detail where amino acids are labeled and distances are measured. On top is ACE2 and on bottom RBD, showing the wild type structure in beige, the UK variant in light blue and the SA variant in pink.
PyMol video of the molecular dynamics simulation ran for 6 nanoseconds with NAMD. On top is ACE2 with Y41 interacting with bottom RBD with Y501. Aromatic-aromatic interactions are strong interactions therefore this UK variant of SARS-CoV-2 binds better to ACE2 receptors infecting more efficiently the human cells.